Essai thérapeutique chez Biotrial à Rennes
Publié en ligne le 11 octobre 2016 - Santé et médicament -

À Rennes, un homme est mort lors d’un essai de phase 1, essai au cours duquel un produit est administré pour la première fois chez l’Homme. Cet essai était réalisé chez des volontaires sains. Le laboratoire portugais BIAL a étudié chez l’animal une molécule inhibitrice de la FAAH (Fatty Acid Amide Hydrolase) BIA 10-2474 et en a confié l’expérimentation chez l’Homme au laboratoire Biotrial à Rennes.
Résumé des faits
Le protocole
Le protocole de l’étude de phase 1 (voir encadré) chez l’Homme comportait quatre essais successifs.
Le premier essai, Single Ascending Dose (SAD), consistait à inclure huit sujets par palier de dose, six recevant le produit actif et deux un placebo. Chaque sujet recevait une dose unique. Les doses prévues étaient de 0,25 mg, 1,25 mg, 2,5 mg, 5 mg, 10 mg, 20 mg, 40 mg et enfin 100 mg. Au premier palier, deux sujets, l’un recevant le produit actif et l’autre le placebo devaient être traités d’abord, les six autres sujets étant traités 24 heures plus tard. Aux autres paliers, les huit sujets étaient traités simultanément. Le protocole prévoyait la possibilité de poursuivre l’escalade de dose en ajoutant aux huit paliers prévus jusqu’à quatre paliers supplémentaires si la dose maximale tolérée n’était pas atteinte, portant ainsi le nombre de sujets de 64 à 96.
Le deuxième essai prévoyait d’administrer à douze autres sujets une dose unique une fois à jeun et une fois non à jeun à quatorze jours d’intervalle, pour étudier l’interaction du produit avec les aliments. La dose n’était pas précisée dans le protocole qui indiquait seulement que celle-ci serait définie durant l’essai SAD (et MAD – Multiple Ascending Dose – si disponible).
Le troisième essai, Multiple Ascending Dose ou MAD, devait inclure simultanément huit nouveaux sujets par palier de dose, six recevant le produit actif et deux le placebo, comme dans le premier essai, la différence étant que chaque sujet recevait la même dose une fois par jour pendant dix jours. Les doses n’étaient pas précisées dans le protocole qui indiquait seulement qu’elles dépendraient des résultats antérieurs et qu’il y aurait entre quatre et huit paliers de dose.
Le quatrième essai devait inclure vingt personnes recevant chacune successivement pendant dix jours le produit ou un placebo dans un ordre aléatoire, avec un période sans traitement entre les deux, pour une étude de pharmacodynamique. La dose n’était pas précisée.
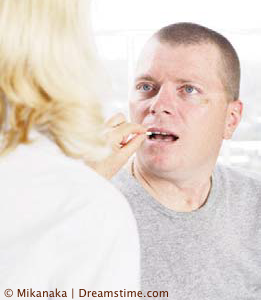
Les essais cliniques qui étudient les effets d’un nouveau médicament chez l’Homme se déroulent dans un ordre précis.
Phase I : étude de l’évolution de la molécule testée dans l’organisme en fonction du temps (cinétique) et analyse de la toxicité sur l’être humain. Cette phase est menée sur un petit nombre de personnes volontaires et non malades.
Phase II :administration du médicament à un petit nombre de patients pour rechercher la plus petite dose efficace et observer des effets secondaires nocifs en utilisant différentes doses.
Phase III : comparaison de l’efficacité du nouveau médicament par rapport au traitement de référence (lorsque celui-ci existe) ou à un placebo (lorsqu’aucun traitement n’existe). Cette phase s’adresse à un grand nombre de patients et dure plusieurs années. Les patients sont sélectionnés sur des critères précis qui permettront de répondre à la question de l’efficacité et du bénéfice du médicament testé comme nouveau traitement standard de la maladie concernée.
C’est à l’issue de ces essais que les autorités sanitaires délivrent l’autorisation de mise sur le marché (AMM). Le nouveau médicament peut être prescrit.
Phase IV : il s’agit du suivi des effets nocifs secondaires des médicaments qui ont été mis sur le marché, cela pour un grand nombre de patients chez qui le nouveau médicament a été prescrit et dans des conditions normales d’utilisation.
Qui autorise les recherches ?
Les procédures administratives réglementaires destinées à autoriser la mise en œuvre d’une recherche clinique et en santé visent avant tout à évaluer la balance bénéfice-risque pour les participants qui se prêteront à cette recherche. Le promoteur de la recherche adresse le protocole de cette dernière à l’autorité administrative compétente, l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) ainsi qu’à un Comité de protection des personnes (CPP) pour vérifier les conditions de sécurité et de protection des personnes participantes. Ces démarches administratives réglementaires aboutissent à une autorisation officielle de la recherche qui peut alors démarrer.
Le protocole a été soumis à l’Agence nationale de sécurité du médicament et des produits de santé (ANSM) et au Comité de protection des personnes (CPP) de Brest, qui l’ont accepté.
La réalisation du protocole
Le premier essai a commencé le 9 juillet 2015. Il a inclus comme prévu huit sujets à chacun des huit paliers de doses, ces dernières allant de 0,25 mg à 100 mg. Le second essai a inclus douze sujets et la dose étudiée a été de 40 mg.
Le troisième essai a inclus huit sujets dans chacun des cinq premiers paliers qui ont été de 2,5 mg, 5 mg, 10 mg, 20 mg et 50 mg. L’état très grave d’un sujet inclus dans ce dernier palier de dose a entraîné l’arrêt des essais. En effet, le dimanche 10 janvier, après avoir reçu la cinquième dose de 50 mg, correspondant à une dose cumulée de 250 mg, dose jamais administrée à l’Homme jusque-là, un volontaire a eu des problèmes de vue, de déglutition et d’élocution qui ont conduit le médecin de Biotrial à l’envoyer aux urgences du CHU de Rennes à 20h30. Dans la nuit et dans la matinée du lundi, son état s’est aggravé et vers midi, il était dans un coma profond. Il décédera une semaine plus tard. En dépit de cette hospitalisation le dimanche soir, le médecin de Biotrial en charge des administrations a délivré une sixième dose aux sept autres sujets à 8 heures du matin le lundi 11 janvier.
Le mardi, un autre sujet a éprouvé des troubles semblables à ceux du premier patient : vision floue et double, puis violents maux de tête. Il a été hospitalisé le mercredi.
Trois autres volontaires, dont les noms n’ont pas été rendus publics, ont eux aussi eu des troubles neurologiques et ont été hospitalisés entre le mercredi et le vendredi. Les IRM cérébrales réalisées à ce moment-là ont montré des lésions. C’est le jeudi que l’ANSM a été prévenue.
Fin février, les volontaires des paliers de dose précédents ont été convoqués au CHU de Rennes pour une IRM de contrôle.
Les enquêtes

Les autorités ont procédé assez rapidement à deux enquêtes administratives, l’une réalisée par un Comité spécialisé scientifique temporaire commandité par l’ANSM, et l’autre par l’Inspection générale des affaires sociales (IGAS), et une information judiciaire pour homicide a été ouverte le 14 juin [1].
Globalement, ces enquêtes administratives ont trouvé assez peu à redire à la conduite des essais. Les experts nommés par l’ANSM ont conclu dans un rapport de 30 pages en ligne le 19 avril [2] que c’était bien le produit qui était en cause. L’enquête de l’IGAS a abouti à un rapport en deux tomes (respectivement de 120 et de 151 pages) [3], incluant une réponse de Biotrial aux critiques (tome 1, pages 85–110 et 117–120, et tome 2, pages 75–146).
Alors que l’IGAS a été missionnée sur l’ensemble du dossier (conditions d’autorisation de l’essai clinique, respect des dispositions de recrutement des volontaires sains, modalités de réalisation de l’essai et de signalement d’événements indésirables graves), ses critiques portent exclusivement sur la gestion de crise. Ainsi, « au terme de ses investigations, la mission estime qu’il n’y a pas lieu de remettre en cause l’autorisation accordée » en faisant toutefois remarquer que « l’économie d’ensemble du protocole et la latitude laissée pour sa mise en œuvre n’offraient pas un cadre suffisant pour la protection des personnes participant à l’essai ». Les trois critiques majeures concernent le comportement de Biotrial lors de l’accident : « ne pas s’être enquis de l’état du sujet hospitalisé lundi matin avant de ré-administrer la molécule » ; « ne pas avoir éclairé, ni recueilli le consentement à poursuivre l’essai des volontaires lundi » ; « ne pas avoir signalé cet effet grave survenu dimanche avant jeudi soir aux autorités de santé » (page 3, synthèse du Tome 1).
Ni l’ANSM ni le CPP de Brest ne sont mis en cause dans le rapport, le protocole et sa validation par les deux organismes étant jugés conformes à la réglementation.
Le Figaro a mis le protocole en ligne (21 janvier 2016), ce que l’ANSM

refusait de faire sous prétexte que c’était la propriété de l’industriel. Devant le fait accompli, l’ANSM a ensuite aussi mis ce protocole en ligne sur son site.
Médiapart a révélé le 24 mai qu’un volontaire du troisième palier de MAD, qui a reçu 10 mg pendant dix jours en novembre 2015, a souffert d’un trouble visuel à deux reprises (en fait, c’est même deux volontaires du palier 10 mg qui ont eu chacun à deux reprises des problèmes de vision, voir rapport du CSST page 20).

L’IRM de contrôle réalisée au CHU de Rennes en février lui a été rendue avec un diagnostic d’accident vasculaire cérébral (AVC) non récent. Le Figaro a soumis cette IRM à quatre experts qui ont conclu que cet AVC s’est produit pendant que le patient prenait la molécule, et a donc probablement un lien avec celle-ci. Depuis, un compte-rendu rectificatif a été envoyé au patient par le CHU de Rennes.
Médiapart a publié le 1er juin le fac-similé de la lettre du médecin de Biotrial envoyée au CHU de Rennes avec le premier patient hospitalisé le dimanche soir. Cette lettre précise que « cette symptomatologie est possiblement en lien avec le produit administré ».
Des questions plus générales
L’affaire de Rennes soulève des questions plus générales quant à la mise en œuvre de tels essais.
1) Pourquoi continue-t-on à administrer simultanément à six personnes un produit nouveau à une dose jamais testée sur l’Homme ? Cette pratique avait pourtant déjà conduit six hommes jeunes en réanimation à Londres en 2006, et le CPP de Marseille en 2006 [4] et l’Agence Européenne du Médicament en 2007 [5] ont recommandé d’inclure les sujets un par un. Malgré cela, l’IGAS et l’ANSM considèrent cette mesure comme optionnelle, son application dépendant de l’évaluation du niveau de risque « pressenti » (rapport IGAS, tome 1, pages 23 et 24), notion non définie. La règle devrait être la prudence, et son non-respect une exception à argumenter.
2) Comment une équipe médicale qui a administré un produit à une personne à une dose jamais testée sur l’Homme, qui a ensuite envoyé cette personne aux urgences et qui, enfin, est responsable de la sécurité de sept autres personnes, peut-elle s’abriter derrière le secret médical (rapport IGAS, tome 1, page 91) pour ne pas s’enquérir de la santé du patient transféré ? Et comment invoquer le fait que le lien de cause à effet n’était pas établi (rapport IGAS, tome 1, page 97 et tome 2, pages 81–87) pour ne pas informer les patients ? Le doute devrait en priorité protéger les volontaires, au lieu de protéger le protocole.
3) Comment l’ANSM et le CPP ont-ils accepté un protocole qui décrivait quatre essais différents en ne précisant les doses que pour le premier de ces essais, laissant ainsi carte blanche aux expérimentateurs pour les trois essais suivants ? Sur quelles bases et par qui ont été déterminées les doses du troisième essai, celui qui a vu les effets graves se manifester ?
4) Pourquoi l’IGAS ne s’est-elle pas procurée le dossier du patient hospitalisé aux urgences de l’hôpital de Rennes le 10 janvier ? Elle semble faire reposer son analyse sur un narratif fourni par Biotrial, y compris une chronologie (rapport IGAS, tome 1, pages 117–120) dans laquelle tous les troubles sont systématiquement omis et remplacés par des points de suspension. Le fac-similé de la lettre du médecin de Biotrial publié par Médiapart permet d’ailleurs de compléter les informations.
Les descriptions des communications entre Biotrial et le CHU de Rennes le dimanche soir sont discordantes. Biotrial affirme avoir reçu un appel leur demandant de reprendre le patient pour la nuit, par manque de place au CHU, ce qui a été interprété comme signalant un état non inquiétant. Le CHU ne mentionne aucune trace de cette conversation, mais le rapport du CHU a été rédigé par l’administration de l’hôpital. Pour justifier la demande de retour du patient le dimanche soir à Biotrial, ce dernier indique que l’hôpital a invoqué l’indisponibilité de l’IRM (rapport IGAS, tome 1, page 89) alors que le site du CHU indique qu’une IRM est dédiée vingt-quatre heures sur vingt-quatre au service des urgences.
5) Pourquoi les deux enquêtes ne se sont-elles pas intéressées au devenir des autres volontaires exposés à six fois 50 mg, ni à celle des sujets exposés aux paliers de dose inférieurs ? Tout ce qu’on sait, c’est que les plus atteints ont été transférés dans des centres de rééducation plus proches de leur domicile, ils se sont ainsi retrouvés soustraits à toute surveillance de la part des autorités censées les protéger.
Indépendance et conflit d’intérêt
Les enquêtes ont été faites par des personnes qui dépendent de l’agence et du ministère des affaires sociales et de la santé, or l’agence, et donc le ministère, sont impliqués dans l’affaire. Les inspecteurs de l’IGAS déclarent s’être appuyés sur l’expertise de l’ANSM et ne trouvent rien à redire au protocole de l’essai qui a été approuvé par l’ANSM.
Conclusion
Les essais de phase 1 posent des problèmes complexes de sécurité et l’évaluation de leur protocole demande des compétences multiples.
À la suite de l’accident de Londres en 2006, l’agence européenne a émis une recommandation insuffisamment claire sur l’échelonnement des inclusions des sujets dans les essais de phase 1, et tous les CPP de France n’ont pas été informés de ce problème. Le protocole de l’essai de phase 1 de BIA 10-2474 était à la fois compliqué et imprécis, mais les autorités ont considéré qu’il était dans les limites acceptables, entérinant ainsi l’inclusion simultanée des sujets dans chaque palier de dose.
La mort d’un volontaire dans un essai de phase 1 est un événement tragique, mais c’est un risque en partie imprévisible ; en général, on retrouvera a posteriori des signaux antérieurs qui, sur le moment, n’avaient pas paru inquiétants. En faire reproche aux investigateurs serait une erreur de jugement. Cependant, Biotrial a failli au devoir de protection des volontaires en administrant une dose supplémentaire aux sept autres sujets après avoir envoyé une personne aux urgences, et les arguties juridiques ou réglementaires invoquées n’arrivent pas à diminuer la responsabilité du laboratoire.
En conclusion, les leçons de cette tragique histoire devraient être
- l’échelonnement systématique de l’inclusion des sujets dans les essais de phase 1
- l’abandon de la notion de « risque identifié » dans les essais de phase 1 : tout événement grave survenant au cours d’un essai de phase 1 doit être, jusqu’à preuve du contraire, considéré comme attribuable au produit testé.
1 | « Essai clinique de Rennes : information judiciaire ouverte pour homicide », Le Monde.fr avec AFP et Reuters, 14 juin 2016.
2 | Rapport du Comité Scientifique Spécialisé Temporaire (CSST) « Inhibiteurs de la FAAH (Fatty Acid Amide Hydrolase) » sur les causes de l’accident survenu à Rennes lors d’un essai clinique de Phase 1 en janvier 2016. Sur archive.org
3 | « Suites de l’accident grave survenu à Rennes : Marisol Touraine renforce la sécurité des volontaires qui participent à des essais cliniques ». Communiqué du 23 mai 2016 accompagnant la mise en ligne des deux tomes du rapport de l’IGAS.
4 | « L’accident de Londres, 13/03/06 », sur le site cpp-sudmed2.fr
5 | European Medicines Agency (Agence européenne du médicament), 19 juillet 2007. « Guideline on strategies to identify and mitigate risks for first in-human clinical trials with investigational medicinal products ». Sur ema.europa.eu
Publié dans le n° 318 de la revue

Partager cet article




L' auteur

Catherine Hill
Catherine Hill est épidémiologiste et biostatisticienne, spécialiste de l’étude de la fréquence et des causes du (...)
Plus d'informationsSanté et médicament

Les cosmétiques : réglementés ? utiles ? toxiques ?
Le 20 décembre 2020
Quelle expertise pour quel usage ?
Le 4 mars 2024
Les prescriptions hors autorisation de mise sur le marché
Le 24 février 2024
Baclofène et alcool : la saga atypique d’un médicament
Le 20 février 2024